 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 来源:国家/各地药监、药审中心,嘉峪检测网编辑整理
【问】单抗下游一次性使用技术的应用及其对应关注点基本有哪些?
【答】一次性使用技术应用于下游生产的多个工序,包括缓冲液配制及贮存、中间产品混匀及贮存、液体转运及过滤、原液贮存及各工序的取样,根据用途的不同,其关注侧重点也不同。
一、一次性搅拌袋、储液袋的技术与安全性关注点如下!
1、完整性要求,破损和漏液问题,对于微生物控制的下游工艺,需尽量避免,应建立使用前检查及使用过程中发生漏液后的相关处理程序。
2、功能性要求,组件的稳固性、搅拌性能、参数测量与调节功能,与支撑容器的适配性/贴合度等。
3、微生物与内毒素控制。
4、化学兼容性与生物相容性。
5、颗粒的脱落。
6、动物源成分。
7、内表面材质,对蛋白的吸附性及稳定性影响。
8、可提取物和浸出物。
9、材料性质,如防冻性(对于原液低温贮存)。
10、特定贮存条件(温度、光照、气体阻隔性等)对于材料的要求。
二、一次性过滤器、深层滤器、除病毒滤器(膜包)的技术与安全性关注点如下:
1、过滤性能要求,如孔径、截留分子量等。
2、过滤器使用条件(压力、流速等)。
3、过滤器完整性测试方法及标准(如涉及)
4、灭菌方式,重复灭菌的性能(如涉及)。
5、微生物与内毒素控制。
6、化学兼容性与生物相容性。
7、颗粒脱落与纤维释放。
8、动物源成分。
9、接触材质对特定检测项目无显著影响(如对残留DNA的吸附等)。
10、可提取物和浸出物。
【问】药包材的等同性研究中,如何判定化学等同?
【答】中国医药包装协会T/CNPPA 3009—2020《药包材变更研究技术指南》明确了“化学等同”的定义为:一般指药包材的可提取物/浸出物谱中未发现提取物/浸出物的种类和含量的增加。
具体研究方法为:采用相同的实验条件(提取和/或模拟提取),对变更前后的药包材进行可提取物谱比较,除不含有机涂层的玻璃和金属类材料药包材外,通常同时满足a)对于有机提取物,大于分析评价阈值(AET)的峰,变更后样品可提取物谱中未出现多余峰,且同时未出现峰面积增高的峰;和b)对于无机元素类化合物,可参照《ICH Q3D元素杂质指导原则》等相关文件,未出现新增元素或元素溶出水平无显著增加,可认为化学等同。
【问】工艺气体终端过滤器有哪些使用要点?
【答】终端滤芯通常安装在使用点之前,以确保进入使用点的气体符合要求。对于无菌制剂,应安装0.22um(更小或相同过滤效力)除菌滤芯,更换前后需要开展滤芯的完整性检测,同时更换前需对其进行灭菌。
对于口服固体制剂,可安装0.22um(更小或相同过滤效力)除菌滤芯,若最终气体质量符合工艺需求,0.45um或1um滤径的过滤器也可用于终端过滤。
制药气体分配系统通常在发生源处使用1um或更小的滤芯进行保护。如果需要无菌气体或无菌排气,通常在交付或使用点由0.22um(更小或相同过滤效力)疏水除菌级滤芯过滤。
无菌制剂与药品或设备内表面直接接触的工艺气体滤芯可以离线灭菌并进行无菌组装,也可以通过纯蒸汽进行在线灭菌(SIP),非最终灭菌制剂建议使用在线蒸汽灭菌方式。对于一次性使用的生物反应器和其他一次性系统,滤芯也可以采用辐射灭菌方式进行灭(PTFE膜经高强度辐射会降低其性能)。
气体除菌滤芯通常由疏水性聚四氟乙烯(PTFE)或疏水性聚偏二氟乙烯(PVDF)制成的0.22um(更小或相同过滤效力)级微孔膜。带有PTFE膜的滤芯与辐射不兼容。一些0.22um(更小或相同过滤效力)微孔膜过滤器也适用于去除小至20nm(0.020um)的空气传播病毒和小至0.003um的空气传播颗粒。
【问】无菌工艺模拟试验中的容器装量有哪些关注点?
【答】灌装体积不必与产品相同,但是灌装体积需要使用与日常生产中相同的方法调整培养基灌装量,应能覆盖到所有容器内表面。适宜的装量需保证产品通过倒置和旋转接触到容器及组件所能接触到的表面,即所有可能直接污染到无菌产品的表面。同时应考虑微生物生长以及培养后观察的需要,灌装量需留足能支持微生物生长的空间和氧气,因此容器不应过度灌装。同时这个量应能够观察到微生物生长产生的浑浊和沉淀现象,能够支持培养基促生长试验和目检的进行。需要特别注意的是,对于无菌粉末分装工艺的模拟,在分装后应采取相应手段,确保所分装的无菌粉末完全溶解于液体培养基中,便于后续观察。
【问】长期未生产品种申请恢复生产时,能否同步提出上市后变更申请?
【答】长期未生产品种申请恢复生产时,如同步发生变更,持有人应参考相关指导原则、根据变更具体情形开展研究和分类,并按规定进行变更管理,如(1)同步发生微小变更的,可自行以年报形式报告,另向省局提出长期未生产恢复生产申请;(2)同步发生中等变更的,可向省局提出长期未生产恢复生产、变更备案申请;(3)同步发生重大变更的,待补充申请获CDE批准后,再向省局提出长期未生产恢复生产申请;(4)同步发生不涉及重大变更的生产场地变更,应参照《浙江省药品上市后变更管理实施细则(试行)》(以下简称实施细则)第十五条相关规定,向省局提出长期未生产恢复生产、生产场地变更、《药品生产许可证》变更(如涉及)、GMP符合性检查(如涉及)申请;(5)同步发生生物制品生产场地变更应按重大变更管理的,应参照实施细则第十六条相关规定,向省局提交《药品生产许可证》变更生产地址和生产范围的申请,获批后针对相关变更事项向CDE提出补充申请,待补充申请获批后,再向省局提出长期未生产恢复生产、《药品生产许可证》变更、GMP符合性检查申请。
【问】长期未生产品种恢复生产同步申请变更时缺少变更前质量研究数据的,应当如何开展相关质量研究?
【答】因药品批准上市后长期未生产等原因,导致变更前的药品质量数据缺失或不具有代表性、可比性,无法开展变更前后质量比对研究的,持有人应按以下原则开展研究:
(一)以法定参比制剂或原研药品为比照对象开展比对研究;
(二)无法定参比制剂和原研药品的,可选择有代表性的市售产品为比照对象开展比对研究,必要时开展临床有效性验证;
(三)无法确定合适比照对象的,应参照国家药监局相关技术指导原则,按照现行药品注册技术要求开展研究,确保药品安全、有效、质量可控。
【问】哪些长期未生产恢复生产品种可以免检查及免注册检验?
【答】长期未生产品种已通过仿制药一致性评价或因处方工艺变更获补充申请批准,且一致性评价和补充申请审评时已安排现场核查和注册检验的,可在恢复生产申请时豁免注册核查和注册检验。
【问】注射器包材供应商发生变更,包材材质和/或类型一致,如何开展等同性研究?
【答】根据中国医药包装协会T/CNPPA 3009—2020《药包材变更研究技术指南》,其研究过程可分为以下几个步骤:
(1)基于先验知识评估风险:若评估相关风险可接受时,可认为等同性及相容性研究完成。(2)若评估仍存在风险,需开展进一步药包材等同性实验研究:①结合药包材质量标准或质量协议,以及药品的性能和生产特性,评估变更前后药包材的保护性和功能性关键性能;②评价药包材变更前后是否化学等同。进行安全性评估:变更前后药包材化学等同,且体外细胞毒性试验可接受(玻璃等无机材料无需进行生物学试验),化学等同性研究结果可用于安全性评估;变更前后的药包材化学不等同,需先定性定量分析新增加的提取物/浸出物,再进行安全性评估(体内、体外生物学评价和可提取物/浸出物的毒理学风险评估);进行相容性研究:化学等同性研究和安全性评估可视为相容性研究的一部分,在此基础上根据风险评估情况判断是否需开展进一步的相容性研究;对药包材的等同性及相容性研究进行总结,确定变更后药包材是否可接受。 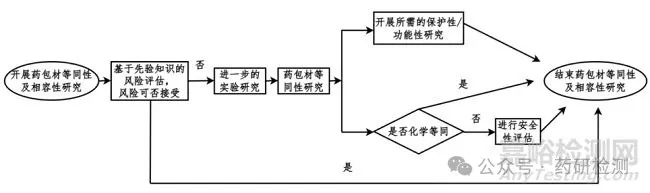
药包材等同性及相容性研究流程图
注:先验知识可能包括:组件配方(如材料组成、添加剂、工艺助剂、催化剂、抗氧剂、色素等)、材料/组件的合规性,如药典或相关标准的符合性、生物反应性试验、TSE/BSE(传播性海绵状脑病/牛海绵状脑病)声明、特殊关注物质无添加声明、化学兼容性、历史提取研究数据及安全性评估数据、组件生产工艺或预处理工艺(如灭菌、清洗、硅化、表面处理)、材料/组件在药品中的使用历史(如已获批上市制剂的处方工艺特性、临床使用信息)、药品处方工艺特性等。
您可以通过扫描下方的二维码关注我们:

【编辑:amanda】 PHEXCOM 本文链接:




 PHEXCOM公众号
PHEXCOM公众号