 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 随着入局者越来越多,Trop2 ADC赛道再次陷入红海竞争。而Trodelvy(戈沙妥珠单抗)的失利,不仅给吉利德带来了难以预估的负面影响,也加重了业内对FDA加速批准含金量的质疑。
近日,吉利德在与FDA协商后,宣布将自愿撤回Trodelvy用于治疗先前已接受过含铂化疗以及PD-1/PD-L1抗体治疗的局部晚期或转移性尿路上皮癌成年患者在美国的加速批准。

此次,吉利德撤回尿路上皮癌适应症的加速批准,与今年5月Trodelvy的一项三期临床试验失利有关。彼时,吉利德宣布Trodelvy在局部晚期或转移性尿路上皮癌的三期确证性TROPiCS-04研究未达主要终点。与化疗相比,Trodelvy未显示出改善患者生存率的效果。安全性方面表现也不尽如人意。
对于本次撤回行为,吉利德表示,该决定将不会影响Trodelvy其他已获批的适应症。不过业内人士认为,Trodelvy新适应症拓展失败对于支持者来说造成了重大打击,面对阿斯利康/第一三共、默沙东等后来者的压力,Trodelvy的首发优势已经逐渐消失殆尽。
与此同时,本次撤回申请中,加速批准(Accelerated Approval,AA)的含金量引发了行业议论。加速批准是FDA在1992年设立的批准新药上市的一种特殊途径,旨在为没有或仅有限治疗选择的严重疾病提供了更快的新治疗方法。然而,行业逐渐发现,近年来被赋予加速批准后又撤回的案例不在少数,这让业内对这一新药上市路径产生了不同角度和程度的争议。
不过,也有行业人士表示,加速批准路径目前仍处于探索阶段,不仅是美国,大量新型治疗模式的涌现也将给全球各地监管部门推动加速批准带来严峻挑战。尽管加速批准的实施可能有不完善之处,但业界应该肯定其对药物开发的总体积极贡献。
TROP2 ADC研发陷入红海
Trodelvy新适应症频频受挫
Trodelvy是全球首个获批上市的Trop2 ADC药物。2020年4月至2021年4月,Trodelvy在一年时间里,接连拿下了三阴乳腺癌(加速批准)、局部晚期/转移性乳腺癌(完全批准)与晚期尿路上皮癌(加速批准)三个适应症。
然而,吉利德大举推进的Trodelvy新适应症研发,随后也开始频频遭遇不利局面。2024年5月,吉利德宣布,TROPiCS-04研究作为膀胱癌条件性批准的确认性试验未能达到预期目标。该药未能在延长接受过PD-1/L1治疗和化疗的膀胱癌患者的生命方面超过化疗效果。
关于这一失败的详细数据,吉利德尚未披露。对此,吉利德表示正在继续分析数据,并将与FDA讨论结果和下一步行动。当时不少业内人士认为,此次试验失败意味着FDA可能会撤销该适应症的批准。
果不其然,5个月过后,经过协商,吉利德自愿撤回Trodelvy用于治疗先前已接受过含铂化疗以及PD-1/PD-L1抗体治疗的局部晚期或转移性尿路上皮癌成年患者在美国的加速批准。对此,吉利德表示,TROPiCS-04研究的更多数据将在即将召开的医学会议上公布。此外,本次新适应症加速批准的撤回不会影响Trodelvy获批的其他适应症。
据了解,加速批准是FDA在1992年设立的批准新药上市的一种特殊途径。即如果在研新药用于治疗严重疾病,FDA可以基于经过验证的可能预测临床获益的合理替代终点或中间临床终点作为审评依据来加速药物的上市批准。2021年,FDA基于2期TROPHY的研究结果,加速批准了吉利德Trodelvy用于尿路上皮癌的后线治疗。
当然,获得加速批准后,申请人仍需要遵守上市后要求(PMR),进行后续的确证性试验,进一步提供药物临床益处的证据,转为完全批准,若确证性试验无法有效证明临床益处,FDA可撤回批准。吉利德进行的3期TROPiCS-04研究,就是为了确认Trodelvy的临床获益,但最终该药在该适应症上还是被判了“死刑”。
事实上,这不是Trodelvy新适应症拓展的第一次受挫。今年1月,吉利德宣布其在预处理NSCLC中的EVOKE-01试验未能在其主要终点OS和PFS上取得成功。这项试验是Trodelvy在NSCLC领域的关键研究,导致了吉利德在4月对Trodelvy的资产价值进行了24亿美元的减值。
这两个试验的失败给Trodelvy蒙上了阴影,同时也削弱了Trodelvy的市场首发优势。目前,Trodelvy正面临阿斯利康/第一三共、默沙东两个强劲的竞争对手的追逐,二者各自有广泛的TROP2 ADC 3期项目正在进行。而国内布局Trop2 ADC的企业更多,主要有科伦博泰、君实生物、多禧生物、映恩生物、迈威生物、百利天恒和恒瑞医药等。
然而,吉利德的高管们对TROP2项目的开发持有长远视角,他们将这一过程比作NBA季后赛,认为尽管遭受初步失利,但比赛还远未结束。这表明吉利德对Trodelvy及其在TROP2靶向治疗领域的长期潜力仍然抱有信心,并准备继续在这一赛道上与其他竞争者展开竞争。
Trodelvy接下来的重要数据公布是EVOKE-03,该试验正在测试吉利德Trodelvy与默沙东的PD-1抑制剂Keytruda(下称K药)在PD-L1高表达的非小细胞肺癌中的联用效果。该试验于2023年2月开始,目前预计的主要完成日期为2027年1月。
业内人士表示,Trop2是继HER2之后ADC领域最为火爆的靶点之一,由于其在多肿瘤中显著高表达,已经吸引来了全球众多药企的布局研究。吉利德能否突破失败阴霾,守住该赛道头把交椅,还有待观察。
10年18项适应症被撤回
加速批准含金量存疑?
据统计,过去十年来,越来越多的创新药通过加速批准途径获得提前上市,其中,超过80%的加速批准资格给到了肿瘤领域。然而,近期发表在JAMA上的一项研究梳理了2013-2023年FDA加速批准的59个抗肿瘤药物的129个适应症,其中仅48个适应症(37%)获得完全批准,63个适应症(49%)尚处于确证性试验阶段,18个适应症(14%)被撤回。
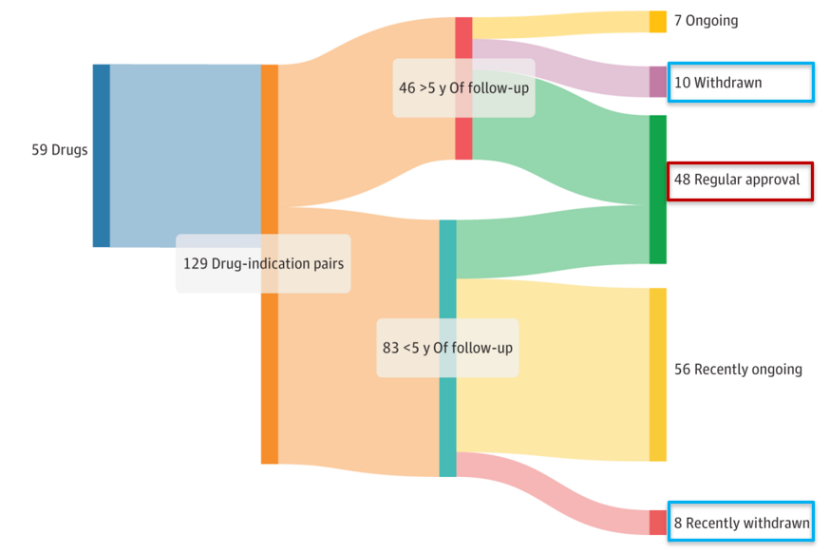
研究者对这些加速审批的撤销进行分析,发现监管方撤销决定包括多种原因,如未能直接重现审批数据、OS数据不达标、审批试验设计不力,以及风险与收益的变化等。与此同时,在具体撤回一款药物的加速批准时,监管方往往要考量更多更复杂的因素以及各个因素之间的相互作用。
以默沙东的K药为例,该药在肝癌适应症上获得加速批准,上市后其三期临床研究Keynote-240失败,但FDA并没有立即取消它的加速批准,这主要是因为FDA之前已经批准了K药针对肝癌的其他上市后临床研究Keynote-394。在这种情况下,FDA通常会优先等待Keynote-394研究结果的出炉,而不会立即做出取消K药加速批准的决定。最终,由于Keynote-394最终达到了临床终点,使得K药针对肝癌的适应症获得了全面批准。
然而,上市后验证性研究未能证实药效却未被撤回,是药物审批和监管领域中一个备受争议的问题。究其原因,FDA或企业可能认为尚有一些患者可能从中获益,或者因为缺乏其他治疗选择。
例如Oncopeptides的多发性骨髓瘤药物 Pepaxto,于2021年2月获得了FDA的加速批准。然而上市后Pepaxto临床研究结果并不如人意,没有达到无进展生存期的主要临床终点。FDA的外部肿瘤学顾问,在2022年9月份的顾问委员会会议上以压倒性多数投票(14:2)反对该药物的获益风险概况。
2023年7月,FDA提议要求Oncopeptides在美国撤回其药品。然而,同年8月,Oncopeptides对FDA的快速撤回提案提出了上诉。在上诉书中,Oncopeptpides猛烈抨击了FDA对试验结果的解释,声称FDA的分析“在科学上不正确”。最终在2024年2月,FDA正式撤回了对于Pepaxto的加速审批决定。
FDA通过7个多月的时间,证明了拥有更多可用治疗选择的愿望不能成为唯一的决定因素。FDA相关负责人表示,撤回Pepaxto的决定可能会让没有其他选择的患者感到不安。然而,FDA也相信患者应该得到其批准的安全有效的治疗。
《JAMA Oncology》上发表的一项研究也发现,从2016年到2022年,超过四分之一的晚期胃癌、乳腺癌、肺癌、肝癌和膀胱癌患者使用了加速批准药物,但后来证明这些药物对这些特定用途无效,甚至有些患者可能已经受到副作用的影响而没有任何益处。
但尽管如此,出于利益或临床缺口等方面的考虑,制药企业仍然竭力维持一款药物的供应。如预防早产的药物Makena,2011年获得加速批准,八年后,上市后研究失败。2019年,FDA召开专家咨询委员会,会议以9比7的投票结果,建议撤回对Makena的加速批准。
当时该产品新的制造商Covis Pharma Group竭尽全力维持该药物的上市,拒绝主动撤回加速审批,并对该结果提起了上诉。几经周折,该药上市的第12年,2023年,FDA做出了撤回对Makena加速批准的最终决定,Makena及其仿制药不再获得批准,也不能在州际贸易中合法分销。
FDA始终认为,“未得到满足的需求”并非将一个无法确定疗效的药留在市场上的理由,而且“如果在进行试验时让它继续留在市场上,反而可能会阻碍其他药物的开发”,甚至是阻碍Makena疗效的验证。
从以上这些撤销加速审批的案例中,业界可以感受到加速批准是一把双刃剑,其优势是显著的,可缩短重要药物的研发周期和上市时间,无药可治、临床急需的患者看到了希望并能够提早获益。但问题也显而易见,通过加速批准途径上市的新药,也存在临床疗效证据并不充分的情况。后续的临床研究如果面临研究结果不及预期的情况,不乏出现难撤回的情况,这对新药研发以及患者获益都会造成负面影响。如何实现加速审批的合理运用,是制药行业与监管机构需持续探索的议题。
【编辑:amanda】 国际药物制剂网 本文链接: http://www.phexcom.cn/hydt.aspx




 PHEXCOM公众号
PHEXCOM公众号