 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 在过去的几十年里,美国FDA通过孤儿药、快速通道、加速批准、优先审评和突破性疗法五种特殊的审查程序来加速满足未被满足的医疗需求,旨在促进和加快针对严重疾病的药物开发。
加速批准使FDA能够用产生临床获益的替代终点来批准治疗未被满足的临床需求的药物。替代终点可以简单理解为异常的生物标志物恢复正常,目前常见的替代指标有药代动力学指标、体外检测指标、影像学指标、疾病生物标志物等。然后获得加速批准药物需要在批准后进行进一步临床试验来确认这些药物是否真正可使患者临床获益。
在近日举办的2024药物信息大会暨展览会上,石药集团肿瘤领域首席医学官黄薇,强生创新制药临床开发部血液和眼科负责人张红,泰格医药高级副总裁、首席医学官陈霞,默沙东研发临床研究部总监毛一香等与会嘉宾对FDA加速审批的案例进行了分享,现场医药从业人员进行了深入讨论。

10年加速批准案例回顾分析
2024年 4 月 ,一项FDA加速批准抗肿瘤药的临床/监管数据研究(Clinical Benefit and Regulatory Outcomes of Cancer Drugs Receiving Accelerated Approval)发布于JAMA的文章中,哈佛大学研究者分析了2013年至2023年期间获得加速批准的129个癌症药物适应证。(涉及59个药物)
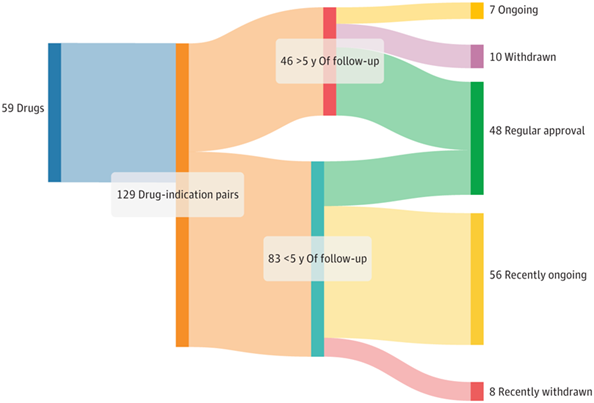
2013年至2023年肿瘤药物加速获批监管结果
根据文章,研究者进行了两项研究:一是着眼于随访超过五年的药物适应证的总生存率和生活质量的改善;二是研究了已转换为常规批准的药物适应证的验证性试验统计数据。
具体而言,129个药物适应证中,46 个随访时间超过 5 年的适应症(2013—2017年批准)。其中,29个(63%)转为常规批准,10个(22%)被撤回,7个(15%)在加速批准后的中位6.3年仍未有明确结局。
在29个转为常规批准的适应证中,只有不到一半(20/46,43%)的20个(69%)证实有临床获益,7个(24%)显示了总生存期(OS)和生活质量(QOL)的改善,7个(24%)改善了OS但未改善QOL,6个(21%)改善了QOL但未改善OS。其余9个(31%)虽然“转正”,但其确证性试验既未显示OS获益也未显示QOL获益。
从时间上来看,加速批准转为常规批准最常发生在1至2年内,撤回批准均匀分布在1到5年之间。时间变化上,退出时间从2013年的9.9年缩短至2017年的3.6年,常规批准的时长从2013年的1.6年增加到2017年的3.6年。
泰格医药高级副总裁、首席医学官陈霞表示,由此可见,转正未必都有临床获益,临床获益未必是生存期和生活质量的双改善。这也凸显了临床获益与监管结局的不确定性。
在第二项研究中,48个药物适应证获得了最终批准(37%),其中,19 个(40%)基于OS改善,21 个(44%)基于PFS或EFS改善,5 个(10%)基于ORR和DOR,2 个(4 %) 基于ORR, 1个 (2%)未显示任何临床获益,仍然获得了常规批准。
此外,还可以看到,2020年只能用OS/PFS转正,2020年后其他指标也可用;2021年—2023年期间的19个AA适应证中有7个(37%)基于ORR或者含ORR指标转正。
通过替代终点来进行加速批准虽然加速了新药上市,但显而易见的是,也对正确评估药物的风险和收益构成了不小的挑战。FDA对此也做出了要求,根据加速批准获批上市的药物需要进行上市后试验。但是关于验证性研究是否应使用替代指标作为主要终点的争议一直未断。加速批准药物的临床获益仍具有很大程度的不确定性,但是对于尚未被满足的临床需求而言,一种新药加速批准上市或许就能带来转机。
加速批准促进更多患者可及
随着新药研发所需要的时间周期越来越长,为满足患者尚未被满足的临床需求,有关监管部门及医药企业一直在寻找更多可能与方法,将有效药物尽早带到临床上。
强生创新制药临床开发部血液和眼科负责人张红对强生多发性骨髓瘤领域双抗产品FDA加速审批进行了分享。她表示,多发性骨髓瘤虽然一直都有新药涌现,但它仍然是一个不断复发且无法治愈的恶性炎症,仍然存在很多未满足的医疗需求。
强生两款治疗多发性骨髓瘤的双特异性抗体均获得FDA突破性疗法认证,为尽快推向临床,强生选择了申请加速批准程序。例如两款药物之一Teclistamab,在2021年申请BTD,2021年6月获得突破性疗法认定,2021年12月进行BLA滚动递交,最终在2022年10月加速批准既往接受过4线治疗的复发难治多发性骨髓瘤患者。
值得注意的是,FDA也对通过加速审批的药物进行了上市后要求。比如对Teclistamab上市后要求开展一项随机对照临床试验,要求具有足够数量的少数种族人群的美国受试者以及老年受试者;以Teclistamab为基础的治疗和SOC进行对比;以PFS为主要重点,OS、ORR、DOR为次要终点。
除了上市后要求,还有上市后承诺,需要在后续的过程中进一步确证接受Teclistamab治疗后神经毒性的发生率、级别和潜在的相关因素;完成Tec-1试验,进一步确证其有效性,获得0RR和DOR;以及CMC(化学成分生产和控制)相关。
默沙东研发临床研究部总监毛一香则分享了默沙东PD-1抗体在肝细胞癌(HCC)二线治疗中的一波三折的加速批准过程。
据悉,肝癌是最常见的原发性肝癌,在全世界男性和女性中分别排名第五和第七位。也是全球第二大与癌症有关的最常见死因,也是美国癌症相关死亡上升最快的死因。然而,肝癌药物研发的失败率极高。美国FDA直到2007年才批准上市肝癌的首个一线靶向药物索拉非尼,后又经过十多年才在2018年批准上市肝癌的第2个一线靶向药物仑伐替尼。
默沙东的两款药物opdivo、keytruda(O药、K药)分别于2017年、2018年获得美国FDA的加速批准,用于经过索拉非尼治疗的肝癌患者。前者在后续的确证性临床III期试验CheckMate-459中,主要终点OS没有达到统计学意义,被FDA肿瘤咨询委员会以5:4反对了加速批准。后者在验证性试验KEYNOTE-240中,主要终点OS和PFS均没有达到,但获得了截然不同的结果,FDA肿瘤咨询委员会以8:0的投票比暂时同意保持K药在二线肝细胞癌的上市状态。
毛一香讲到,给两款药物带来不同命运的是FDA“悬空”加速批准(dangling Accelerated Approval)这一程序。“悬空”加速批准用于描述确证性试验没有证实临床获益,但上市许可仍在继续批准。在某些情况下,如果存在持续未满足的临床需求,并且存在确证性试验不能验证临床获益的特殊原因,在另一项确证性试验完成时,该适应症仍可上市销售。
加速批准机制的意义在于加速将新药物推向市场,以满足临床需求。通过加速批准程序,企业能更早上市产品,使患者能够更早获得新的治疗选择。抓住加速批准机会,在早期研究设计等阶段,对研究终点、研究数据要求、研究设计不确定性等方面尽早与监管部门进行沟通,做好万全准备。
【编辑:amanda】 国际药物制剂网 本文链接: http://www.phexcom.cn/hydt.aspx




 PHEXCOM公众号
PHEXCOM公众号