 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 经历一波三折,礼来的阿尔茨海默病(AD)创新药Kisunla(通用名donanemab)终于在7月初获美国FDA批准上市,成为第二款上市的AD新药。Kisunla与卫材/渤健的Leqembi(通用名lecanemab)跨越了AD新药研发的“死亡之谷”,一定程度上证明淀粉样蛋白假说有效性,使AD患者有望拥有更多治疗选择。但跨越研发“死亡谷”后,AD新药还有众多难关需要克服。
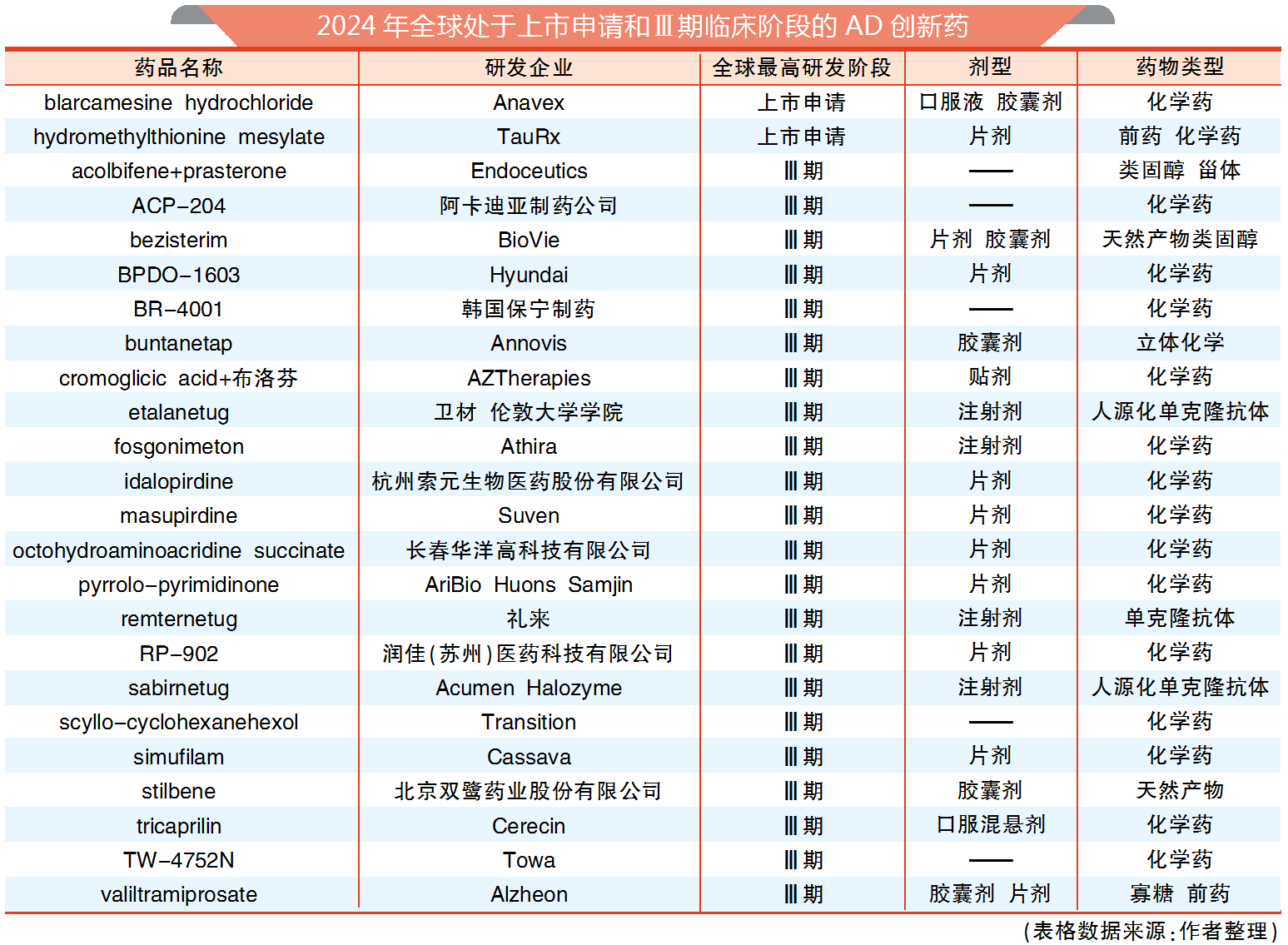
失意者铺前路
AD发病机制至今有待探索,目前主流假说包括β淀粉样蛋白(Aβ)异常沉积、tau蛋白磷酸化、胆碱能损伤等。渤健的Aduhelm(通用名aducanumab)于2021年获FDA加速批准,头顶“首款AD疗法”的光环,但其药物真实益处、试验设计的透明性和数据解释的一致性均备受质疑,上市后争议不断,如今已撤市。
对于AD新药研发,已有众多药企遭遇滑铁卢。美国药品研究与制造商协会(PhRMA)的报告显示,1998-2017年,AD药物研发成功率仅为2.7%。在Aduhelm获批上市前,已经有4款治疗AD的抗Aβ单抗在Ⅲ期研发阶段折戟:辉瑞和强生的bapineuzumab、礼来的solanezumab、罗氏的crenezumab和罗氏的gantenerumab。
其他靶点的AD研发药物失败的例子也不少,如β-分泌酶1(BACE1)小分子抑制剂类药物:默沙东的verubecestat、阿斯利康和礼来联合开发的lanabecestat及强生的atabecestat均在Ⅲ期临床试验阶段失败。
但这些失意者也为后来的研发之路提供方向。bapineuzumab的研究使业界认识到抗Aβ单抗可以在患者中引起淀粉样蛋白相关影像学异常;solanezumab的研究则使研究人员发现要对入组患者进行淀粉样Aβ水平的检测等。此外,抗Aβ单抗赛道的屡败屡战也让业界认识到,这类产品可能更适合无症状但有AD风险的群体或早期AD患者。
新秀来势汹汹
如今,Leqembi与Kisunla的成功上市标志着继胆碱酯酶抑制剂及NMDA受体拮抗剂之后,抗Aβ单抗步入治疗AD的主流药物队列。
Kisunla的适用患者人群与Leqembi的适用患者人群几乎相同,两种药物在开始治疗前都需要通过PET成像确认患者脑组织中的β淀粉样蛋白病理,并且都有淀粉样蛋白相关成像异常(ARIA)风险的黑框警告。ARIA异常可能表现为脑肿胀和脑出血。
尽管Leqembi已在2023年被FDA批准上市,占据一定先发优势,但Kisunla在获批前就被视作Leqembi的强劲对手。
作为首款在淀粉样蛋白斑块被清除后可以停止使用的药物,Kisunla在给药频率和输注时间方面具有明显优势:其使用方法是每4周进行1次持续0.5小时的静脉输注,而Leqembi则是每2周进行1次持续1小时的静脉输注。
在给药剂量方面,不同于Leqembi,Kisunla的剂量不需要根据患者体重进行调整。
定价方面,礼来将Kisunla定价为695.65美元/瓶,一年的治疗费用为3.2万美元,而Leqembi在美国上市时的定价为2.65万美元/年,但考虑到Kisunla在达到治疗效果后可以不再进行治疗,总体上讲使用Kisunla应该更便宜。
在疗效方面,根据各自被FDA批准上市的关键性临床研究,两者的区别不大。
但在安全性方面,与Leqembi相比,Kisunla的缺点是在淀粉样蛋白相关影像异常方面的安全性较差。Leqembi的标签要求在注射前进行4次脑部磁共振成像扫描,而Kisunla则需要5次。另外,在Kisunla的关键性临床试验中有3例与治疗相关的死亡,而这种现象在Leqembi的关键性临床试验中未发生。
巨头多维争雄
Leqembi与Kisunla上市,只是卫材/渤健和礼来在AD治疗领域激烈竞争的其中一个战场。今年6月,卫材和渤健宣布,其向FDA提交的Leqembi静脉注射剂型的补充生物制品许可申请获FDA受理。这个申请获FDA批准后,患者在第1个月的治疗起始阶段仍需要每2周静脉输注1次,但在后续的维持治疗阶段每月静脉输注1次即可。FDA将其PDUFA日期定为2025年1月25日。
此外,卫材还向FDA递交了第一个月起始阶段后的维持阶段每周皮下注射1次的Leqembi自动注射器的生物制剂许可申请,目前已被FDA给予快速通道指定,这样FDA可对药物申请的部分内容进行滚动审批。若成功获批,患者可直接在家中给药,且注射时间缩短。如此一来,Leqembi将挑战礼来的Kisunla达到疗效后可以停药的给药方案所带来的市场优势。
自然,礼来也积极应对市场挑战,正努力推进在研抗Aβ单抗remternetug研发。较早的临床研究提示,remternetug可能比Kisunla更有效地清除脑内斑块。目前礼来正在开展将静脉输注的remternetug及皮下注射的remternetug与安慰剂对照比对的Ⅲ期临床研究,以评估remternetug对于早期AD患者的安全性和疗效。
笔者预计,此类产品对于有AD认知和功能下降风险,但尚无临床症状人群的AD预防领域会有更为广阔的市场。在此领域,礼来Kisunla的TRAILBLAZER-ALZ 3Ⅲ期临床研究及卫材的AHEAD 3-45Ⅲ期临床研究都正在进行中。
此外,tau蛋白磷酸化假说也是药企研发AD新药的指导理论之一,上述3家巨头也在布局tau靶点AD新药:卫材正在日本、美国及欧洲开展抗微管结合区tau抗体etalanetug针对显性遗传性阿尔茨海默病(DIAD)的Tau NexGenⅡ/Ⅲ期临床研究;渤健启动了针对AD的靶向tau的反义核苷酸疗法BIIB080的Ⅱ期临床研究以及开展O-GlcNACase(OGA)酶小分子抑制剂BIIB113的Ⅰ期临床研究;礼来目前也正在开展OGA小分子抑制剂LY3372689用于AD治疗的Ⅱ期临床研究。
落地困难重重
走通了研发路,只是漫漫长征第一步。从Leqembi上市至今仍困于可及性难题来看,Kisunla上市后要解决的问题并不少。
在今年6月举行的2024年美国核医学与分子影像学会年会(SNMMI)上,许多专家围绕AD新药进行讨论。
梅奥诊所的Mary Ellen Koran指出,即使在专业的失智中心,AD误诊率也超过25%。犹他大学放射学和影像科学系主任Satoshi Minoshima则进一步补充道,美国PET成像仪容量有限、放射科医生和认知障碍专家短缺等客观原因限制了此类AD新药可及性。
Leqembi在美获批已接近1年,但根据《纽约时报》对梅奥诊所、麻省总医院和加州大学旧金山分校医学中心的神经科医生进行的调研显示,符合Leqembi治疗条件的美国患者不到20%。患者需接受PET成像或腰椎穿刺检查,且要进行MRI检查,筛查其他脑部疾病等。由于担心副作用,患者还被要求对APOE4基因变体进行基因检测,拥有两个APOE4基因变体的患者可能被拒之门外。此外,PET成像费用高昂,患者还需考虑医保覆盖问题。
虽道阻且长,所幸创新者已迈出持续前进的步伐。根据Market.us的数据,2023年全球AD治疗药物销售额为55亿美元,并将以18.8%的年复合增长率递增,到2033年达到308亿美元。期待更多创新者踏上征服AD之路。
【编辑:amanda】 国际药物制剂网 本文链接: http://www.phexcom.cn/hydt.aspx




 PHEXCOM公众号
PHEXCOM公众号