 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 HER3靶点一直是肿瘤赛道聚焦的潜力领域,临床探索的一举一动都牵动着行业敏感的神经。
日前,BioNTech发布公告,公司合作伙伴MediLink(宜联生物)收到FDA的通知,要求部分暂停正在进行的BNT326/YL202的I期试验(NCT05653752),暂停新受试者入组。这是因为在试验过程中已有受试者因药物副作用而死亡,FDA对受试者面临重大疾病或伤害风险表示担忧。
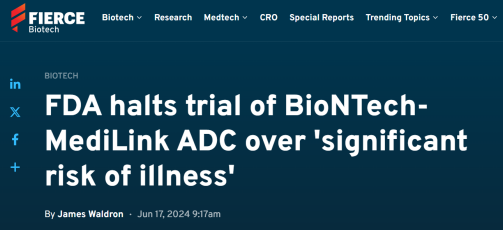
FDA要求审查临床和安全数据,与该机构共享可用的药理学数据,并在研究者手册中提供有关安全发现的额外信息,包括在研究YL202-INT-101-01和YL202-CN-201-01中观察到的5级不良事件,即死亡事件。

在全球HER3 ADC领域的竞争格局中,第一三共的HER3-DXd已经提交了上市申请。国内的创新企业在这一领域也表现活跃,百利天恒、恒瑞医药、信达生物、映恩生物等公司都取得了进展,凭借出色的技术平台开拓了海外市场。
靶向HER3的ADC已经吸引着众多制药企业涌入赛道争夺市场先机。此次早期临床高剂量探索引发的安全性事件,HER3 ADC临床探索道路将发生怎样的变化?
剂量探索引发监管关注
事实上,YL202的研发进程处于全球领先地位。
在临床前实验中,这款HER3靶向ADC能够有效地将细胞毒性药物传递到表达HER3的肿瘤细胞中,同时对健康组织的毒性影响较小。目前全球尚未有任何上市的针对HER3的疗法,这表明该靶点具有巨大的潜力。

值得一提的是,宜联生物在刚刚结束的ASCO 2024年会上公布了HER3-ADC YL202-INT-01-01试验的初步积极结果。BioNTech也公布了临床试验NCT05653752的结果:在52名入组患者中,疾病控制率(DCR)达到了94.2%,客观缓解率(ORR)达到了42.3%。同时,BioNTech在报告摘要中还表示,该研究“显示出足够的安全性和耐受性”。
在早期临床试验过程中,ADC产品剂量爬坡阶段观察产品安全性,也是临床试验必须走过的道路。
信息显示,截至2月4日,共有54名患者接受了BNT326/YL202的I期试验治疗。在第5剂量组(4mpk)中,有2名患者死亡;而在第7剂量组(5.5mpk)中,有1名患者死亡。此外,在6月4日的一份报告中,BioNTech表示,在治疗期间共有14人死亡,其中3人死于与药物相关的不良反应。
目前,第一三共和阿斯利康的HER3-ADC(patritumab Dxd)是在治疗EGFR突变的后线非小细胞肺癌患者中取得最先进展的药物。该药物已于去年底提交了BLA申请,并在今年6月底进行了PDUFA评审。根据数据显示,该药物的治疗效果约为30%的客观缓解率(ORR)和大约半年的中位无进展生存期(mPFS)。
对于patritumab Dxd的BLA数据,涉及225名EGFR突变后经过治疗的患者,有65%的患者出现了大于等于三级不良事件(TEAE)。patritumab Dxd的停药比例为7%,而有20%的患者出现了三级及以上的TEAE(如中性粒细胞减少、血小板减少),其中有一例患者出现了五级ILD间质性肺炎。
目前,YL202的临床和安全性数据正在接受FDA的审查。而据最新的媒体消息,BioNTech 在SEC 公告中表示,I期剂量探索阶段发生的高剂量风险事件。在基于已有数据的情况下,BioNTech与宜联生物正在主动跟美国FDA更新安全性数据,以便进一步进行数据分析。
全球研发竞争如火如荼
国内外企业正在探索HER3这一新兴靶点带来的广阔市场机会。
去年年底,第一三共/默沙东宣布美国FDA受理了他们开发的HER3 ADC候选产品HER3-DXd(U3-1402)的生物制品许可申请,并授予其优先审评资格,用于治疗既往接受过两种或两种以上全身治疗的局部晚期或转移性EGFR突变非小细胞肺癌(NSCLC)成人患者。FDA的审评决议日期(PDUFA日期)定于2024年6月26日。HER3-DXd有望成为全球首个上市的、针对HER3靶点的创新疗法。
近日,Endeavor BioMedicines宣布成功完成了1.325亿美元的C轮融资,获得资金将用于推动其治疗特发性肺纤维化(IPF)和进行性肺纤维化(PPF)的在研小分子疗法ENV-101的临床开发。此外,这笔资金还将支持针对HER3阳性实体瘤的ENV-501临床概念验证研究。ENV-501是新一代靶向HER3的ADC,结合了先进的化学和遗传学特性,旨在优化药物递送至肿瘤,从而提高其效力并减少非靶向毒性。Endeavor计划在2024年启动ENV-501的1/2期临床试验。
业内人士认为,随着临床开发的不断深入,ADC药物在多种癌症中都显示出良好的抗癌效果,有望改变各种癌症的治疗策略。而HER3的表达因主要见于肺癌、乳腺癌、结直肠癌、胃癌等多个常见肿瘤,具有成为泛癌ADC的潜力,正受到制药企业的研发热捧。
根据智慧芽新药情报库,在全球范围内,在研的HER3 ADC多达25款,涉及220条研发状态。其中,本土制药企业的创新表现尤为活跃,不乏恒瑞医药、百利天恒、信达生物、宜联生物、映恩生物等先锋药企的身影。
百利天恒全资子公司SystImmune开发的BL-B01D1是全球首创的EGFR/HER3 ADC,目前临床开发进度仅次于第一三共和默沙东的HER3-DXd。BL-B01D1可靶向在大多数上皮性肿瘤中高度表达的EGFR和HER3,由SystImmune 独家拥有的双特异性抗体和“连接子-有效载荷”组成,其中包含稳定的、可切割的连接子和拓扑异构酶抑制剂。
DB-1310是映恩生物基于其DITAC技术平台开发的靶向HER3的ADC产品,由新型人源化抗Her3单克隆抗体通过马来酰亚胺可裂解linker与专有的DNA拓扑异构酶I抑制剂共价连接,DAR值约为8。2023年5月,DB-1310的临床试验申请获CDE受理,拟用于治疗晚期/转移性实体瘤。根据2023 AACR公布的数据,DB-1310表现出优于U3-1402的肿瘤抑制效果和安全性;并且在与EGFR小分子抑制剂联合治疗中表现出优秀的协调治疗效果。
今年开年,恒瑞医药对外公告称,其自主研发的HER3 ADC创新药注射用SHR-A2009获得美国FDA授予快速通道资格,用于治疗经第三代EGFR-TKI和含铂化疗后疾病进展的EGFR突变的转移性NSCLC。根据公告,SHR-A2009也是恒瑞医药历史上第一个获得美国FDA快速通道资格认定的创新药。
无独有偶,今年2月,信达生物开发的HER3 ADC IBI133也在国内首次申报临床。据悉,IBI133是信达生物布局的一款HER3 ADC,已于去年12月在澳大利亚启动了一项I/II期临床试验(NCT06170190),针对实体瘤患者。
HER3 ADC研发竞争,究竟谁能够率先冲线,仍需时间给出答案。HER3靶点一直是肿瘤赛道聚焦的潜力领域,临床探索的一举一动都牵动着行业敏感的神经。
日前,BioNTech发布公告,公司合作伙伴MediLink(宜联生物)收到FDA的通知,要求部分暂停正在进行的BNT326/YL202的I期试验(NCT05653752),暂停新受试者入组。这是因为在试验过程中已有受试者因药物副作用而死亡,FDA对受试者面临重大疾病或伤害风险表示担忧。
FDA要求审查临床和安全数据,与该机构共享可用的药理学数据,并在研究者手册中提供有关安全发现的额外信息,包括在研究YL202-INT-101-01和YL202-CN-201-01中观察到的5级不良事件,即死亡事件。
在全球HER3 ADC领域的竞争格局中,第一三共的HER3-DXd已经提交了上市申请。国内的创新企业在这一领域也表现活跃,百利天恒、恒瑞医药、信达生物、映恩生物等公司都取得了进展,凭借出色的技术平台开拓了海外市场。
靶向HER3的ADC已经吸引着众多制药企业涌入赛道争夺市场先机。此次早期临床高剂量探索引发的安全性事件,HER3 ADC临床探索道路将发生怎样的变化?
剂量探索引发监管关注
事实上,YL202的研发进程处于全球领先地位。
在临床前实验中,这款HER3靶向ADC能够有效地将细胞毒性药物传递到表达HER3的肿瘤细胞中,同时对健康组织的毒性影响较小。目前全球尚未有任何上市的针对HER3的疗法,这表明该靶点具有巨大的潜力。
值得一提的是,宜联生物在刚刚结束的ASCO 2024年会上公布了HER3-ADC YL202-INT-01-01试验的初步积极结果。BioNTech也公布了临床试验NCT05653752的结果:在52名入组患者中,疾病控制率(DCR)达到了94.2%,客观缓解率(ORR)达到了42.3%。同时,BioNTech在报告摘要中还表示,该研究“显示出足够的安全性和耐受性”。
在早期临床试验过程中,ADC产品剂量爬坡阶段观察产品安全性,也是临床试验必须走过的道路。
信息显示,截至2月4日,共有54名患者接受了BNT326/YL202的I期试验治疗。在第5剂量组(4mpk)中,有2名患者死亡;而在第7剂量组(5.5mpk)中,有1名患者死亡。此外,在6月4日的一份报告中,BioNTech表示,在治疗期间共有14人死亡,其中3人死于与药物相关的不良反应。
目前,第一三共和阿斯利康的HER3-ADC(patritumab Dxd)是在治疗EGFR突变的后线非小细胞肺癌患者中取得最先进展的药物。该药物已于去年底提交了BLA申请,并在今年6月底进行了PDUFA评审。根据数据显示,该药物的治疗效果约为30%的客观缓解率(ORR)和大约半年的中位无进展生存期(mPFS)。
对于patritumab Dxd的BLA数据,涉及225名EGFR突变后经过治疗的患者,有65%的患者出现了大于等于三级不良事件(TEAE)。patritumab Dxd的停药比例为7%,而有20%的患者出现了三级及以上的TEAE(如中性粒细胞减少、血小板减少),其中有一例患者出现了五级ILD间质性肺炎。
目前,YL202的临床和安全性数据正在接受FDA的审查。而据最新的媒体消息,BioNTech 在SEC 公告中表示,I期剂量探索阶段发生的高剂量风险事件。在基于已有数据的情况下,BioNTech与宜联生物正在主动跟美国FDA更新安全性数据,以便进一步进行数据分析。
全球研发竞争如火如荼
国内外企业正在探索HER3这一新兴靶点带来的广阔市场机会。
去年年底,第一三共/默沙东宣布美国FDA受理了他们开发的HER3 ADC候选产品HER3-DXd(U3-1402)的生物制品许可申请,并授予其优先审评资格,用于治疗既往接受过两种或两种以上全身治疗的局部晚期或转移性EGFR突变非小细胞肺癌(NSCLC)成人患者。FDA的审评决议日期(PDUFA日期)定于2024年6月26日。HER3-DXd有望成为全球首个上市的、针对HER3靶点的创新疗法。
近日,Endeavor BioMedicines宣布成功完成了1.325亿美元的C轮融资,获得资金将用于推动其治疗特发性肺纤维化(IPF)和进行性肺纤维化(PPF)的在研小分子疗法ENV-101的临床开发。此外,这笔资金还将支持针对HER3阳性实体瘤的ENV-501临床概念验证研究。ENV-501是新一代靶向HER3的ADC,结合了先进的化学和遗传学特性,旨在优化药物递送至肿瘤,从而提高其效力并减少非靶向毒性。Endeavor计划在2024年启动ENV-501的1/2期临床试验。
业内人士认为,随着临床开发的不断深入,ADC药物在多种癌症中都显示出良好的抗癌效果,有望改变各种癌症的治疗策略。而HER3的表达因主要见于肺癌、乳腺癌、结直肠癌、胃癌等多个常见肿瘤,具有成为泛癌ADC的潜力,正受到制药企业的研发热捧。
根据智慧芽新药情报库,在全球范围内,在研的HER3 ADC多达25款,涉及220条研发状态。其中,本土制药企业的创新表现尤为活跃,不乏恒瑞医药、百利天恒、信达生物、宜联生物、映恩生物等先锋药企的身影。
百利天恒全资子公司SystImmune开发的BL-B01D1是全球首创的EGFR/HER3 ADC,目前临床开发进度仅次于第一三共和默沙东的HER3-DXd。BL-B01D1可靶向在大多数上皮性肿瘤中高度表达的EGFR和HER3,由SystImmune 独家拥有的双特异性抗体和“连接子-有效载荷”组成,其中包含稳定的、可切割的连接子和拓扑异构酶抑制剂。
DB-1310是映恩生物基于其DITAC技术平台开发的靶向HER3的ADC产品,由新型人源化抗Her3单克隆抗体通过马来酰亚胺可裂解linker与专有的DNA拓扑异构酶I抑制剂共价连接,DAR值约为8。2023年5月,DB-1310的临床试验申请获CDE受理,拟用于治疗晚期/转移性实体瘤。根据2023 AACR公布的数据,DB-1310表现出优于U3-1402的肿瘤抑制效果和安全性;并且在与EGFR小分子抑制剂联合治疗中表现出优秀的协调治疗效果。
今年开年,恒瑞医药对外公告称,其自主研发的HER3 ADC创新药注射用SHR-A2009获得美国FDA授予快速通道资格,用于治疗经第三代EGFR-TKI和含铂化疗后疾病进展的EGFR突变的转移性NSCLC。根据公告,SHR-A2009也是恒瑞医药历史上第一个获得美国FDA快速通道资格认定的创新药。
无独有偶,今年2月,信达生物开发的HER3 ADC IBI133也在国内首次申报临床。据悉,IBI133是信达生物布局的一款HER3 ADC,已于去年12月在澳大利亚启动了一项I/II期临床试验(NCT06170190),针对实体瘤患者。
HER3 ADC研发竞争,究竟谁能够率先冲线,仍需时间给出答案。
【编辑:amanda】 国际药物制剂网 本文链接: http://www.phexcom.cn/hydt.aspx




 PHEXCOM公众号
PHEXCOM公众号