 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务  图1.《关于发布境内生产药品再注册申报程序和申报资料要求的通告(征求意见稿)》
图1.《关于发布境内生产药品再注册申报程序和申报资料要求的通告(征求意见稿)》
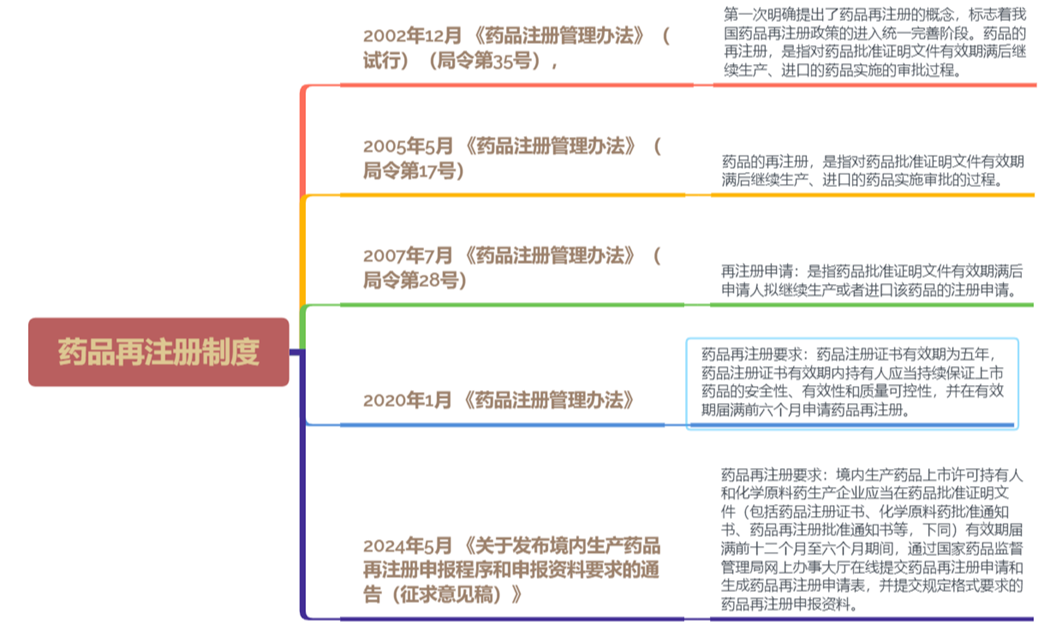 图2.药品再注册制度变化和完善梳理
图2.药品再注册制度变化和完善梳理





【编辑:amanda】 国际药物制剂网 本文链接: http://www.phexcom.cn/hydt.aspx




 PHEXCOM公众号
PHEXCOM公众号