 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 临床试验数据质量是评价新药有效性和安全性的关键,完整数据链贯穿临床试验全过程。数据溯源是对数据的追本溯源,以实现对历史数据的重现。
2023年5月17日,广东省药学会公众号广东药物临床试验发布《药物临床试验 源数据管理·广东共识(2023版征求意见稿)》,供各医疗机构和临床试验相关方参考。新版《药物临床试验质量管理规范》(2020版)(2020年第57号)自2020年7月1日起施行,对临床试验的管理带来新挑战和新问题,该指南对临床试验中涉及源数据/源文件的管理要求提出了一些业内认可的做法和建议,明确源数据、源文件的概念、类别以及载体形式,统一申办方/CRO、研究者、试验机构管理人员等各方对源数据管理的认识差异,提高临床试验数据管理的质量。
PART.01
源文件/源数据/元数据
你能分清吗?
在药物临床试验的日常工作中,我们几乎每天都在和临床试验中的文件与数据打交道,了解清楚源文件/源数据/元数据的概念可以使工作更加得心应手。
源文件(Source Documents)
GCP定义:指临床试验中产生的原始记录、文件和数据,如医院病历、医学图像、实验室记录、备忘录、受试者日记或者评估表、发药记录、仪器自动记录的数据、缩微胶片、照相底片、磁介质、X光片、受试者文件,药房、实验室和医技部门保存的临床试验相关的文件和记录,包括核证副本等。源文件包括源数据,可以以纸质或者电子等形式的载体存在。(来源:2020年7月1日实施的《药物临床试验质量管理规范》(2020年第57号))。
广东共识版定义:是指包括了源数据的文件,可以是原始文件或其核证副本,以纸质或者电子等形式存在。比如医院病历、医学图像、实验室记录、备忘录、受试者日记或者评估表、发药记录、仪器自动记录的数据、缩微胶片、照相底片、磁介质、X光片、受试者文件,药房、实验室和医技部门保存的临床试验相关的文件和记录(CRF中的数据不是源数据,通常不作为源文件)。(来源:药物临床试验 源数据管理·广东共识(2023版征求意见稿))
源数据(Source Data)
GCP定义:指临床试验中的原始记录或者其复印件(即核证副本,指经过审核验证,确认与原件的内容和结构等均相同的复制件,该复制件是经审核人签署姓名和日期,或者是由已验证过的系统直接生成,可以以纸质或者电子等形式的载体存在。核证副本也可以作为有效的记录)上记载的所有信息,包括临床发现、观测结果以及用于重建和评价临床试验所需要的其他相关活动记录。
广东共识版定义:指临床试验中的原始记录或其核证副本上记载的所有信息,包括临床发现、观测结果以及用于重建和评价临床试验所需要的其他相关活动记录。源数据应当具有可归因性、易读性、同时性、原始性、准确性、完整性、一致性和持久性。(来源:药物临床试验 源数据管理·广东共识(2023版征求意见稿))
元数据(Meta Data)
是用来定义和描述数据的数据,通过定义和描述数据,可以支持对其所描述的数据对象的定位、查询、交换、追踪、访问控制、评价和保存等诸多管理工作。(来源:2020年12月1日实施的《药品记录与数据管理要求(试行)》)
PART.02
药物临床试验中
源数据和源文件管理
在临床试验中,源数据是作为溯源依据的数据,源文件是承载源数据的原始文件,自开展临床试验数据现场核查以来,药监管理部门不断强调临床试验数据的产生、收集、记录和报告过程应真实、完整和准确。临床试验中立项、伦理、过程中以及结题等,都离不开源数据管理,而这些源数据管理都符合相关要求吗?
源数据的类别和管理要求
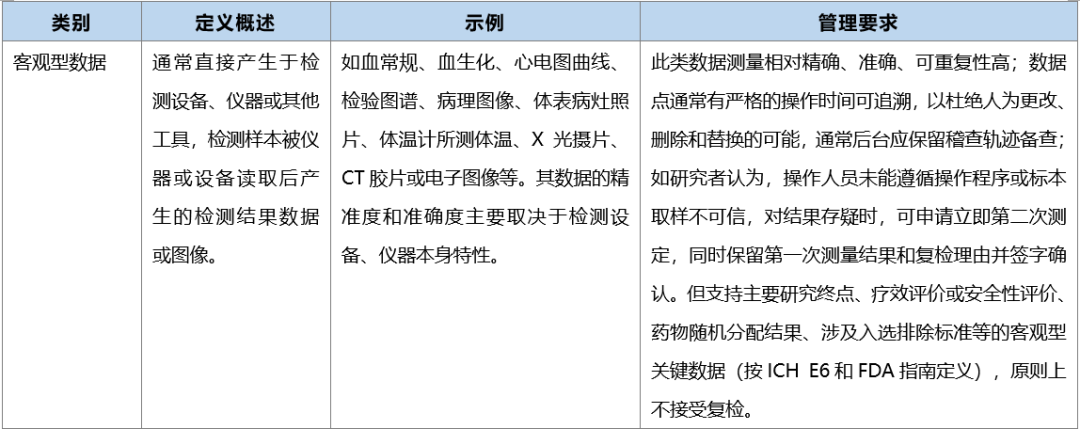

源文件的类别和管理要求
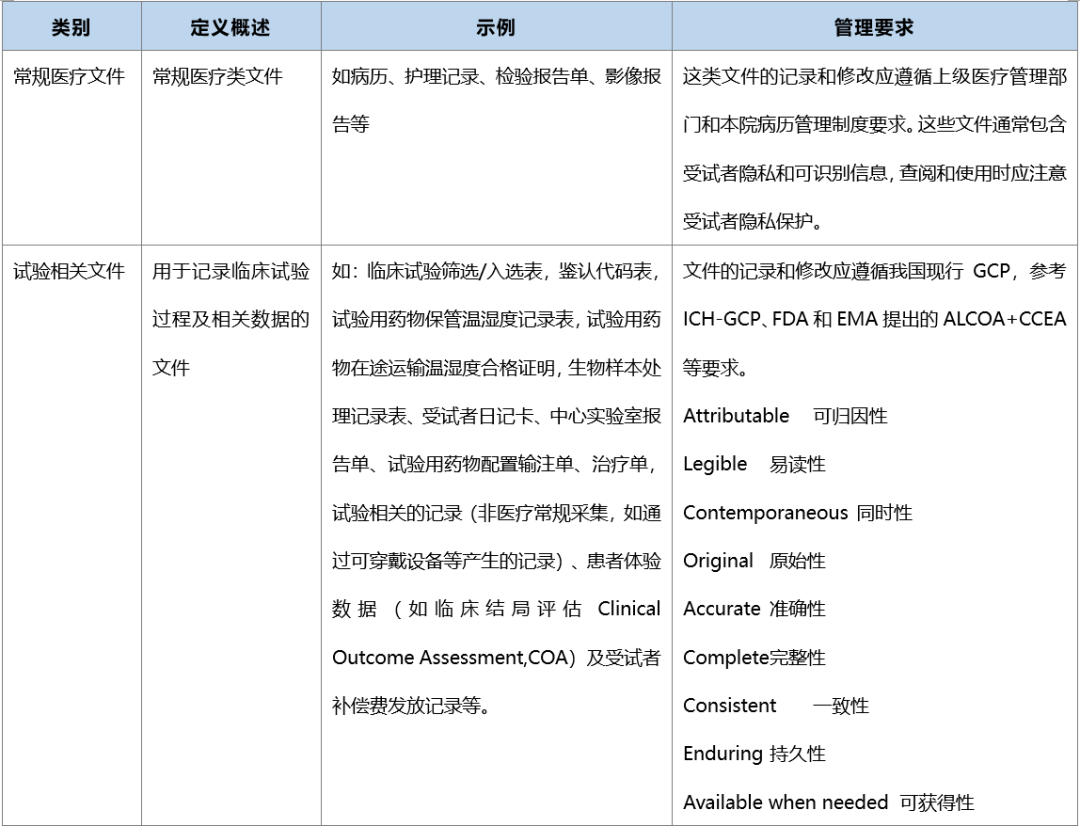
源数据如何鉴定?
在临床试验中,同一事件的数据可能会由不同人获取并记录,如临床护士和研究医生可能都会采集试验中受试者的生命体征,并分别记录在护理单和病历中,或由同一人获得并记录在不同的地方,如研究人员将采集的生命体征分别记录在临床病历和受试者文件中,为确证病例报告表(CRF)所记录数据的有效来源,建议在临床试验开始前,采用“源数据鉴认表”,源数据鉴认表可参见以下范例。
表.临床试验-源数据鉴认表建议范例
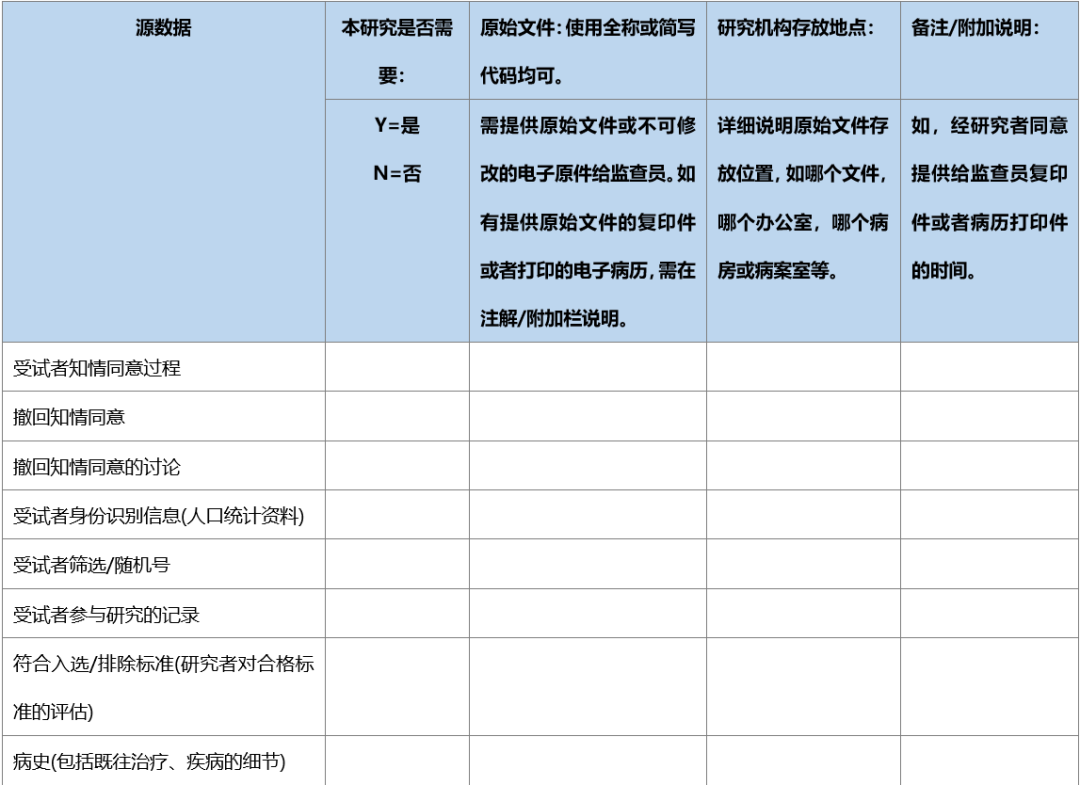
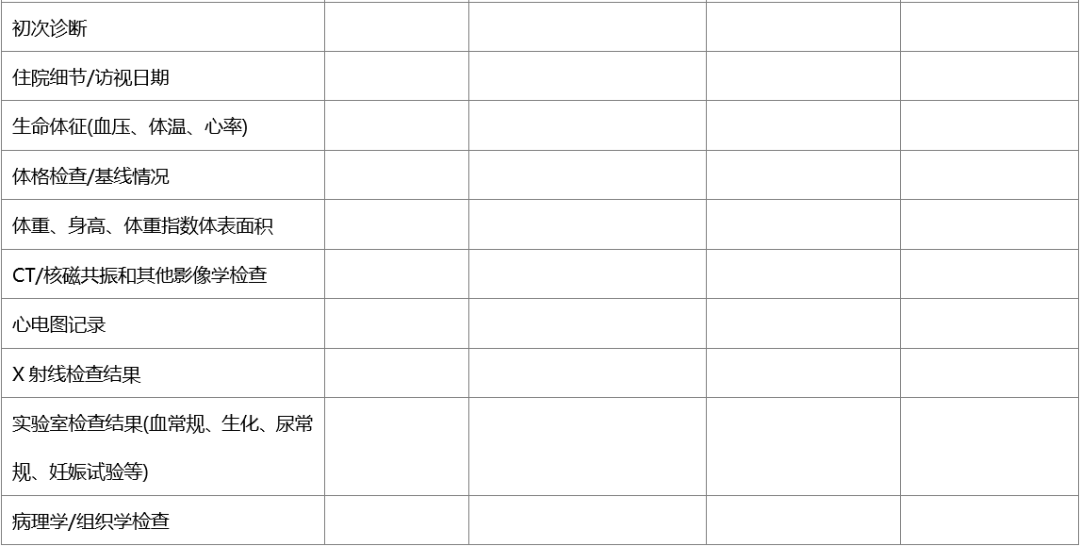
本研究整个持续时间内,如果原始文件的存放位置发生变化,监查员、研究者或指定人员需更新此表,或需要用新版本表格记录发生变化的信息,要保留所有既往版本,并标记为“被取代”。

PART.03
临床试验核查中
源数据管理监管关注点
国家药品监督管理局颁布的《药品注册核查要点与判定原则(药物临床试验)(试行)》(2022年1月1日实施)是药品临床试验项目数据核查的金标准。检查要点包含2个部分11大项:
药物临床试验-临床试验部分(8大项)
1) 临床试验许可与条件
2) 伦理审查
3) 临床试验实施过程
4) 试验用药品管理
5) 生物样品管理
6) 中心实验室及独立评估机构
7) 临床试验数据采集与管理
8) 委托研究
药物临床试验-生物样品分析部分(3大项)
9) 生物样品分析条件与合规性
10) 生物样品分析实验的实施
11) 记录的管理
《药品注册核查要点与判定原则(药物临床试验)(试行)》对研究过程中原始记录和数据进行核实、实地确认,经核查确认发现以下情形之一的,核查认定为“不通过”,11种情形如下:
编造或者无合理解释地修改受试者信息以及试验数据、试验记录、试验药物信息;
• 以参比制剂替代试验制剂、以试验制剂替代参比制剂或者以市场购买药品替代自行研制的试验用药品,以及以其他方式使用虚假试验用药品;
• 隐瞒试验数据,无合理解释地弃用试验数据,以其他方式违反试验方案选择性使用试验数据;
• 瞒报可疑且非预期严重不良反应;
• 瞒报试验方案禁用的合并药物;
• 故意损毁、隐匿临床试验数据或者数据存储介质;
• 关键研究活动、数据无法溯源;
• 申报资料与原始记录不一致且影响结果评价;
• 其他严重数据可靠性问题;
• 拒绝、不配合核查,导致无法继续进行现场核查;
• 法律法规规定的其他不应当通过的情形。
参考资料:
1.www.sinopharmacy.com.cn
专栏作者
滴水司南
生物医药高级工程师、执业药师,自媒体时代的药政法规搬运工,坚定终身学习的目标,坚持学用结合,努力做到知行合一。
如果这篇文章侵犯了您的权利,请联系我们。
【编辑:amanda】 国际药物制剂网 本文链接: http://www.phexcom.cn/hydt.aspx




 PHEXCOM公众号
PHEXCOM公众号