 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 前言:在CFDA的GMP飞检中,出现了一些典型的数据可靠性问题。在药品研制现场考核、生产现场检查中,这些问题也同样具有启示作用,必须引起注意,对有些不符合规定的做法应立即整改。
1. 中心化验室中高效液相色谱仪(型号:岛津SPD-10A、编号:QC-002)的计算机日志显示该公司存在修改系统时间的行为。
分析:计算机的系统时间不可以修改,应采用必要措施保证不被修改,同时对实验人员加强培训。
2. 空白批记录未受控发放。
分析:GMP范围内所有的空白记录,如批生产记录、批检验记录都必须受控,不可以随意打印。研发实验室中的记录可以不必受控。
3. 高效液相色谱仪(编号:QC042)及FTIR650傅里叶变换红外光谱仪(编号:QC006)未对人员权限进行分级,计算机系统时间可修改;高效液相色谱仪(编号:DC002)仅设置开机密码,且为三人公用。
分析:HPLC、IR必须进行权限管理,系统时间不可以修改,每个人都应使用各自的密码,不可以共用,且密码应定期更换。
4. 高效液相色谱仪(HPLC)、傅里叶变换红外光谱仪所用计算机及工作站未合理设置登录权限,部分仪器未开启审计追踪功能并修改日志记录。编号DJS001的HPLC未开启审计追踪功能,且存在多次修改时间的日志记录,编号DJS046的HPLC工作站审计追踪报告显示,其工作时间先后顺序为2017年6月21日、2017年3月3日、2017年3月24日、2017年1月14日、2017年6月17日。
分析:HPLC、IR建立权限管理后,应开启审计追踪,打印的图谱应带有审计追踪信息。
5.7月2日现场检查时,脱包间(房间号:01-01-22)查见低硼硅玻璃安瓿的物料状态标识上当日发放记录未及时填写,物料状态标识数量与现场实物不一致。安瓿物料状态标识中的初始量123.72万支、《安瓿传递窗使用记录》中显示安瓿使用量约117.6万支及现场剩余量19万支,三者数量不平衡。
分析:要注意账、物、卡相对应,发放时应及时填写记录,物料标识要清楚,数量准确,物料流向清楚。
6. 灯检已结束,《灯检工段原始记录》未及时记录,操作人员已签名,但灯检数、不合格数等均未填写,且无法从灯检机上获得相关数据。
分析:检验记录应及时记录,不能造假,必要时对人员进行培训,仪器设备应与检验项目相匹配,不合适的仪器应立即换掉。
7. 洗瓶批生产记录中洗瓶工序要求填写洗瓶槽内水温度50-60℃,记录中填写为具体温度值,如54℃,但现场未见温度测量设备。员工表述该温度为手触摸洗瓶槽外温估计所得。
分析:每个操作单元、工序中应配备相应的检测设备,并经过校验合格,在有效期内,不应根据人的感官去判断是否符合要求。
8. 检验报告中的图谱时间、峰面积与电子数据不一致。151203批炎可宁片盐酸小檗碱含量测定色谱图上的实验时间为2016年4月15日,样品峰面积分别为118.5001、117.2649、117.0647、117.3108。检查高效液相色谱(设备编号:A0605011)电子数据发现2016年4月15日未进行检验,但在2016年3月15日、2016年4月13日的检验序列中发现该批的产品的检验电子数据,其中3月15日序列中平行两份样实验数据峰面积为117.4199、117.2649、117.0647、116.5474,4月13日序列中平行两份样实验数据峰面积为124.4098、125.8490、6.9355、6.6652。高效液相色谱仪(设备编号:A0605013)中发现删除命名为炎可宁数据文件的痕迹。实验室Lei KA DM500显微镜无测微尺,但提供的检验记录中有显微特征尺寸。
分析:严谨进行图谱套用,数据录入时要保证正确无误,且与图谱一致。严禁删除数据和图谱,检测设备必须经过校验合格,处于正常使用时间范围内。
9. 现场检查发现企业正在编造批量为48万袋的复方板蓝根颗粒(15g)工艺验证方案及报告。
分析:在现场检查时,可以对现场环境进行检查,如发现有其他品种在编造数据、不按GMP要求进行操作等行为,同样会被记录下来。
10. 存在删除图谱现象,且未记录原因。恢复该计算机系统回收站已清空的数据发现,有三张名称分别为“对01(20160930 17:16:22).hw;样01(20160930 17:36:42).hw;样02(20160930 17:56:00).hw”的图谱创建时间分别为2016年9月30日的17:16:38、17:55:57、18:13:54,这些时间点介于三七伤药片成品(批号:160912、160913)相关检验图谱创建时间、修改时间之间;红外分光光度计(型号FTIR-650,编号133001)的计算机系统回收站有乙醇160502(食用)、乙醇160702(食用)等7个批次的图谱,与保存的用于物料放行的同一名称的图谱不一致。
分析:检查时检查组拥有数据“恢复软件”!可以恢复回收站中已清空的数据!要小心!
11. 2017年3月10日上午抽查了化验室1号高效液相色谱仪计算机系统显示时间为2017年4月10日。计算机系统使用日志中日期时间有修改,化验室负责人解释为因部分图谱丢失,为了保证重新打印的图谱与当时监测报告的打印时间一致,对电脑时间进行了修改。
分析:当需要对图谱进行重新打印时,不可以随意修改电脑时间,应按照实际时间进行打印并说明原因。
12. 检查发现,企业用于申报生产的9个批次的人血白蛋白长期稳定性考察3个月、6个月、加速试验6个月大部分铝离子实际检测结果高于药典规定的不得高于200µg/L的标准,与注册申报数据不符。
分析:药品注册申报资料中的数据要与记录保持一致,要认真核对,避免出现不一致的情况。
13. 分析仪器管理混乱,部分仪器缺少使用记录。企业未执行计算机化系统附录相关要求,仪器工作站均未设置权限,未分级管理,数据未定期异地备份和存档,存在新数据覆盖旧数据的现象,不能溯源。
分析:精密分析仪器应具有仪器使用记录。应执行GMP计算机化系统附录规定,按要求制定文件、培训,数据应定期异地备份和存档,保证数据安全,严禁数据覆盖,所有的数据都应该能够溯源。
14. 白芍(批号YL24411607-1、检验单号YL2441-B048)【含量测定】对照品1高效液相色谱图显示进样时间为2016年7月11日 10:57:00,打印时间为9:49:45;对照品2高效液相色谱图显示进样时间为2016年7月11日 11:25:49,打印时间为9:50:48。样品1高效液相色谱图显示进样时间为2016年7月11日 11:59:47,打印时间为9:51:46;样品2高效液相色谱图显示进样时间为2016年7月11日 12:51:44,打印时间为9:52:13。
分析:图谱的真实性问题在审评时直接不批准。
15. 炒白芍(批号1607244151、检验单号CP2441-B049)【含量测定】样品1高效液相色谱图显示进样时间为2016年7月14日 13:27:39,样品2高效液相色谱图显示进样时间为2016年7月14日 14:01:07,该批批生产记录中炮炙岗位生产记录显示,“操作起止时间:7月14日 15:20—17:30”;筛选岗位生产记录显示,操作生产时间自7月14日 17:00开始, 18:00结束;包装岗位生产记录显示,操作时间为7月14日17:30开始,18:40结束。询问(质量受权人)说,一般情况下,成品检验从包装间抽取样品。
分析:样品的检测时间、生产时间、取样时间冲突,暴露出了真实性问题。
16. 煅磁石含量测定需使用重铬酸钾滴定液(0.01667mol/L)滴定:批号:1604041151 (检验单号:CP0411-B009,检验时间:2016.4.5)检验原始记录中显示该滴定液的浓度为0.01667mol/L;批号:1505041151 (检验单号:CP0411-B088,检验时间:2016.5.17)检验原始记录中显示该滴定液的浓度为0.01625mol/L,未提供上述两份滴定液的配制记录。
分析:滴定液的配制记录、标定记录是容量法检查的重点。
17. 煅炉(型号:WDL-15,设备编号:SS-09)的《主要设备运行记录》上记录的信息与实际生产情况不相符:
分析:设备运行记录要与批生产记录中的时间点相对应,如果出现矛盾,说明存在问题。
总结:通过以上问题分析,可见实验室数据可靠性方面的缺陷主要集中在以下几方面:
1、仪器设备的权限管理、审计追踪、数据备份、系统时间修改;
2、记录受控、编造、不一致的情况;
3、图谱套用;
4、试验操作不规范;
5、实验室管理不规范等。
2017年9月19日,审核查验中心官网发布了《MHRA发布2016年GMP检查趋势分析》,公布了一份2016年GMP检查缺陷数据趋势报告,该报告反映,2016年检查发现缺陷中出现频率最高的前10类的数量见表1。
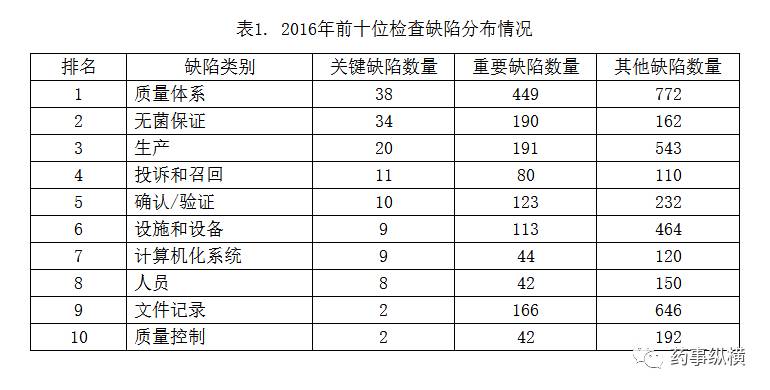
从表中发现,计算机化系统、人员、记录、质量控制这些可能涉及数据可靠性的方面排在前十位中,企业可以对照上述案例和缺陷分布情况,开展自我评估,并作为自检和持续改进的一部分。
此外, 2017年9月13日,审核查验中心官网发布了《TGA发布关于数据可靠性的政策声明》,该文件建议制药商在数据管理和数据可靠性方面关注以下几方面:
1、审查现有质量体系程序和系统,以确保数据可靠性得以维持。重点关注:
*纸质复印文件和批记录的控制,包括空白表单和模板的控制和使用
*电子数据和审计追踪的访问、生成、控制和审查流程
*电子系统的验证
*GMP 数据的存储、备份和归档系统
*员工培训和对数据可靠性的认识
2、通过自查/内部审计计划持续审查数据可靠性控制的有效性。
3、通过供应商管理和审计计划审查关键服务提供商的数据完整性控制。
TGA 希望企业在自查出数据可靠性问题时,应通过更新质量体系、设备和软件以解决所识别出的问题。
作者认为,企业在药品研发环节也应该加强GMP合规性意识,结合国内外法规要求对研发实验室进行管理和完善,重视数据可靠性方面的问题,尽早采用预防措施。
【编辑:amanda】 国际药物制剂网 本文链接: http://www.phexcom.cn/hydt.aspx




 PHEXCOM公众号
PHEXCOM公众号